Hírek
2026.02.18 12:26
2026.02.18 12:22
2026.02.17 12:37
2025.03.19
2025.02.28
2024.12.20
2024.05.22
2024.05.20
2024.05.15 19:34
Fórum
Látogató számláló
0
1
4
8
8
2
2
Anyagcserebetegségek - rövid összegzés
Szerző: Katóné J. Zsuzsa
Az MPS Társaság az MPS betegeket és egyéb tárolási betegséggel küzdő betegeket és családjaikat fogja össze.
Ezekben a betegségekben közös, hogy mind anyagcserebetegségek és lizoszomális tárolási betegségek, erről kicsit később bővebben olvashatunk. Először nézzük át a betegség okát.
A betegséget egy genetikai hiba okozza, amely többnyire autoszomális recesszív öröklődésmenetet mutat, kivéve az MPS II-t, amely X kromoszómához kötötten öröklődik.
Lássuk, mit jelent az autoszomális recesszív öröklődésmenet:
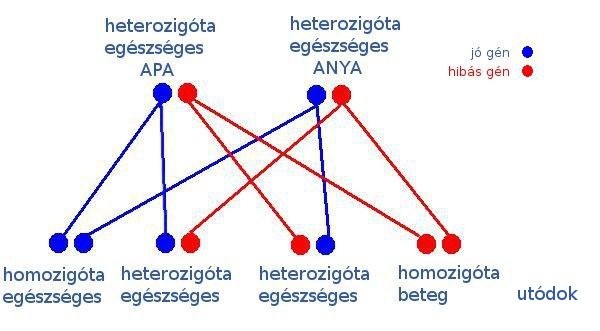
Adott egy egészséges apa és egy egészséges anya, akik nem is sejtik, hogy mindketten ugyanazt a hibás gént hordozzák. 25 % az esélye annak, hogy mindketten egészséges gént adjanak tovább gyermeküknek, ebben az esetben az utód egészséges lesz és még csak nem is hordozza a betegséget. 50 % az esélye annak, hogy egyikük az egészséges, másikuk a hibás gént adja át a gyermekének, ebben az esetben az utód egészséges lesz, csak épp olyan hordozó, mint a szülei. Ekkor sem derül fény arra, hogy bárki is hordozza a családban a hibás gént. Azonban 25 % esélye van annak, hogy mindketten a hibás gént fogják továbbadni gyermeküknek, ekkor az utód beteg lesz, függetlenül attól, hogy fiú vagy lány.
Most tekintsük át, mit jelent az X kromoszómához kötődő öröklődésmenet:
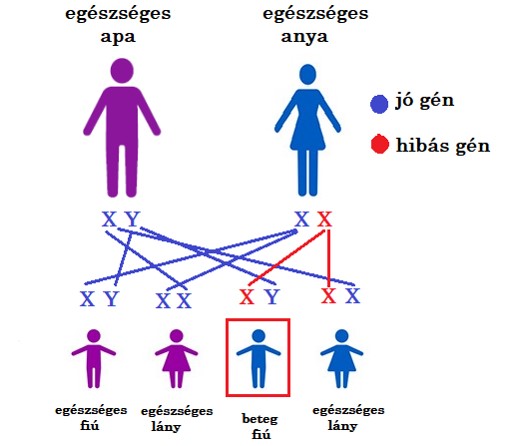
Adott egy egészséges apa és egy egészséges anya. Az anya a nemi kromoszómán egy hibás gént hordoz, de erről nincs tudomása, mert ő egészséges, hiszen van egy egészséges X kromoszómapárja is.
Amennyiben lányuk születik, az utód mindenképpen egészséges lesz, de 50 % az esélye annak, hogy hordozza a hibás gént és 50 % az esélye annak, hogy még csak nem is hordozó.
Amennyiben a párnak fia születik, 50 % az esélye annak, hogy beteg lesz. A gyermek az apától mindenképpen egészséges Y gént kap, az anyától viszont 50 % valószínűséggel egészséges, 50 % valószínűséggel beteg gén érkezik. Ez utóbbi esetben a fiú beteg lesz, hiszen nincs az X kromoszóma mellett egy egészséges másik X kormoszómapár. Láthatjuk, hogy elegendő 1 hibás gén ahhoz, hogy a fiú beteg legyen! Ebben az esetben csak és kizárólag fiúk lesznek betegek.
Most térjünk rá arra, hogy mit jelent az anyagcserebetegség és a lizoszomális tárolási betegség.
Az anyagcsere az a folyamat, amelynek során a szervezet az elfogyasztott táplálékokot az emésztőrendszerben kis egységekre bontja, abból kinyeri a működéséhez szükséges összetevőket, a már nem hasznosítható anyagokat pedig eltávolítja. Anyagcserebetegség akkor jön létre, ha ebbe a folyamatba hiba csúszik. Az anyagcserebetegségeknek rengetek típusa van, ezek közül sok gyógyítható és megfelelő táplálkozással kezelhető (például a cukorbetegség, a laktózintolerancia, stb.), néhány azonban gyógyíthatatlan.
Miért van az, hogy valamire van gyógymód, valamire pedig nincs? Azért, mert az egyik betegség gyakori, a befektetőknek az évek során „érdemes” volt pénzt fektetni a kutatásokba, sok beteg volt, akiken kikísérletezték a gyógyszert, így létrejött a hatékony orvosság. A másik betegség pedig ritka, nem versengenek a befektetők azért, hogy beszálljanak a kutatásokba, beteg sincs annyi, hogy 1-1 gyógyszert tesztelni tudjanak, így ezekre a betegségekre nincs még gyógymód. Az MPS és a társaságunk által összefogott betegségek ez utóbbi csoportba tartoznak. A magyar élő betegek száma kb. 25 fő, ami többnyire állandó, mert folyamatosan születnek beteg gyermekek, de körülbelül ugyanannyit el is veszítünk közülük.
A lizoszóma a citoplazmában elhelyezkedő eukarióta sejtszervecske, amelynek alapvető jelentősége van a sejt védekezési mechanizmusaiban és bizonyos anyagcsere-folyamatokban. Emiatt a sejtek kémiai gyárának is nevezhetjük, ugyanis rengeteg enzimet termelnek. Ha a fent említett hibás gén miatt akár csak 1 db lizoszomális enzim (vagy transzporter) nem termelődik a szervezetben vagy rendellenesen működik, lizoszomális tárolási betegség jön létre.
A hiányzó enzimtől függően a különböző anyagok (lipidek, szacharidok, stb.) nem bomlanak le a sejtekben, hanem felhalmozódnak a lizoszómákban. Ezek a bomlástermékek alapesetben nem lennének toxikusak, a gondot az jelenti, hogy nem tudnak tovább bomlani, így egyre több és több halmozódik fel belőlük a sejtben az évek során. Ennek következtében a lizoszómák kitágulnak, a sejtek deformálódnak, nem tudják ellátni a feladatukat, így a sejtek által felépített szervek is károsodni fognak. A károsodás valamennyi szervet érint, mint például a tüdőt, szívet, májat, csontokat, agyat, bőrt, stb. Ennek kívülről is látható tünetei jelentkeznek a betegeken: fizikai deformitások, fogyatékosság, neurológiai elváltozások formájában. Ezek az elváltozások súlyosak és visszafordíthatatlanok.
Előfordul, hogy az enzim az elvártnál alacsonyabb szinten működik, vagy egyáltalán nem termelődik, a betegség lefolyása ennek függvényében alakul. Az enzim működésének szintje enzimaktivitás vizsgálattal mutatható ki. Ha nagyon alacsony az enzimaktivitás, a betegség első jelei születés után hamar megjelennek és a betegség hamar manifesztálódik, ha magasabb az enzimszint, több év is eltelik, mire észrevehetők lesznek a tünetek.
Mukopoliszacharidózis
A mukopoliszacharidózis tehát egy enzimhiányból eredő anyagcserebetegség, egy lizoszomális tárolási betegség. A beteg szervezetében a mukopoliszacharidok (szakirodalom többféleképpen írja: mucopolysaccharid/mucopolisacharid, de glükózaminoglikánnak, GAG-nak is nevezi, az oldalon is többféleképpen szerepelhet, ami nem elírás, a cikk szerzője által használt változatot jelzi) lebontása egyáltalán nem, vagy nem tökéletesen megy végbe, így kóros mértékben felgyülemlik a sejtekben.
Az emberi szervezet kötőszövetet tartalmaz (porc, ín és bőr), amely a csontokkal együtt egy szilárd vázat (csontváz) és borítást (bőr) képez. A kötőszövet egyik fontos alkotóeleme a mukopoliszacharid (MPS), amely cukormolekulák láncolatából tevődik össze. Az MPS-ek állandóan lebomlanak és szintetikusan újból előállnak; a lebomlás a lizoszómákban történik enzimek segítségével. Az enzimek feladata az MPS-ek meghatározott sorrendben kis alkotórészekre való lebontása. A különböző MPS-ek lebontásához több különböző enzimre van szükség. Mivel kötőszövet az emberi test minden részében található, a mukopoliszacharidózis betegségeknél az egész test károsodik. Ahogy egyre több lebontatlan anyag halmozódik fel a szervezetben, úgy romlik a beteg állapota. A rosszul lebontott anyagok az évek múlásával a szervezetben felhalmozódnak, egyre több kárt okoznak, a betegek leépülnek, szinte minden szervük károsodik, többnyire nem érik meg a felnőttkort.
A lebomlás zavarai következtében a vizeletben megnövekedett mennyiségben válnak ki MPS-ek, ezt GAG-ürítés formájában mérik az orvosok.